Decision time for UK medical device regulation: diverge or converge with the EU?
Published on 28th March 2024
The MHRA appears to be considering an approach for UK regulations that would broadly mirror the EU model
The start of 2024 brought updates and progress in both the EU and the UK on medical devices regulation. In January, the European Commission issued proposals to address implementation challenges faced by medical devices manufacturers, while a Medicines and Healthcare products Regulatory Agency (MHRA) roadmap outlined UK reforms to deliver greater cooperation with overseas regulators and likely recognition of medical devices that are compliant with EU rules.
The EU Medical Device Regulation
In 2016, the European Union (EU) implemented the EU Medical Device Regulation (MDR) (2017/745) to replace existing directives with the goal of improving safety, transparency and traceability of medical devices within the EU.
The EU MDR was published in May 2017 but did not come into force until 26 May 2021 (after its implementation was delayed due to the Covid-19 pandemic).
Reform package
The EU MDR reforms included a wider definition of "medical device". Some products that had not been regulated as medical devices were brought within the new framework, such as cosmetic contact lenses. Conformity assessment procedures and the risk-based classification system for medical devices were also enhanced, with increased obligations for high-risk devices. Notified bodies responsible for initial conformity assessment of high-risk devices were handed new responsibilities, including to carry out unannounced audits and post-market surveillance.
New requirements were introduced for conducting clinical evaluations and investigations to demonstrate compliance with safety standards and specifications, while manufacturers and operators were given increased responsibilities for post-market surveillance and quality management.
Unique device identification numbers and an enhanced European Database on Medical Devices (EUDAMED) were brought in to facilitate traceability and transparency of devices and related information. The new regulations also aimed to establish closer coordination through information exchanges and coordinated assessments to ensure implementation and international regulatory cooperation.
EU delays and challenges
While progress has been made in implementing the EU MDR, there have been a host of significant delays in transitioning medical devices to the new rules. These delays and challenges are due to factors such as a shortage of notified bodies and lack of capacity for device certification, supply chain issues leading to shortages of raw materials in the EU, and manufacturers not being prepared to comply with the new regulations.
In order to prevent further disruptions to the supply and availability of medical devices in the EU and enhance safety and transparency, the Commission has proposed giving more time to manufacturers to comply with the EU MDR.
New timeframe proposals
The new proposals include the extension of the transition periods for manufacturers to comply with the new rules – high-risk devices would have until December 2027, medium-risk devices would have until December 2028, and lower-risk devices until December 2029.
A phased roll-out of the implementation of EUDAMED is also proposed to facilitate effective implementation of the EU MDR.
Mandatory prior notice for supply interruptions at least six months in advance would also be required from manufacturers. Competent authorities, distributors and healthcare providers would need to be notified if they anticipate a disruption or discontinuation in the supply of medical devices.
The reaction to the proposals have been positive. MedTech Europe has emphasised the importance of extended transition periods and the mandatory use of EUDAMED.
UK medical devices regulatory framework
The UK was a member of the EU when the EU MDR was drafted and had actively participated in the shaping of the regulations before Brexit. However, the delayed implementation of the EU MDR meant that it did not form part of the UK's "retained EU law" at the end of the Brexit transition period, which concluded on 31 December 2020.
The MHRA launched a consultation in 2021 on future UK medical device regulations. In January 2024, it released the roadmap, which outlined future reform, to provide clarity and preparation opportunities for medical device providers and the industry as a whole. The MHRA aims to learn from the challenges faced during the implementation of the EU MDR regime in the EU.
The EU MDR and UK timeline: 1993-2030
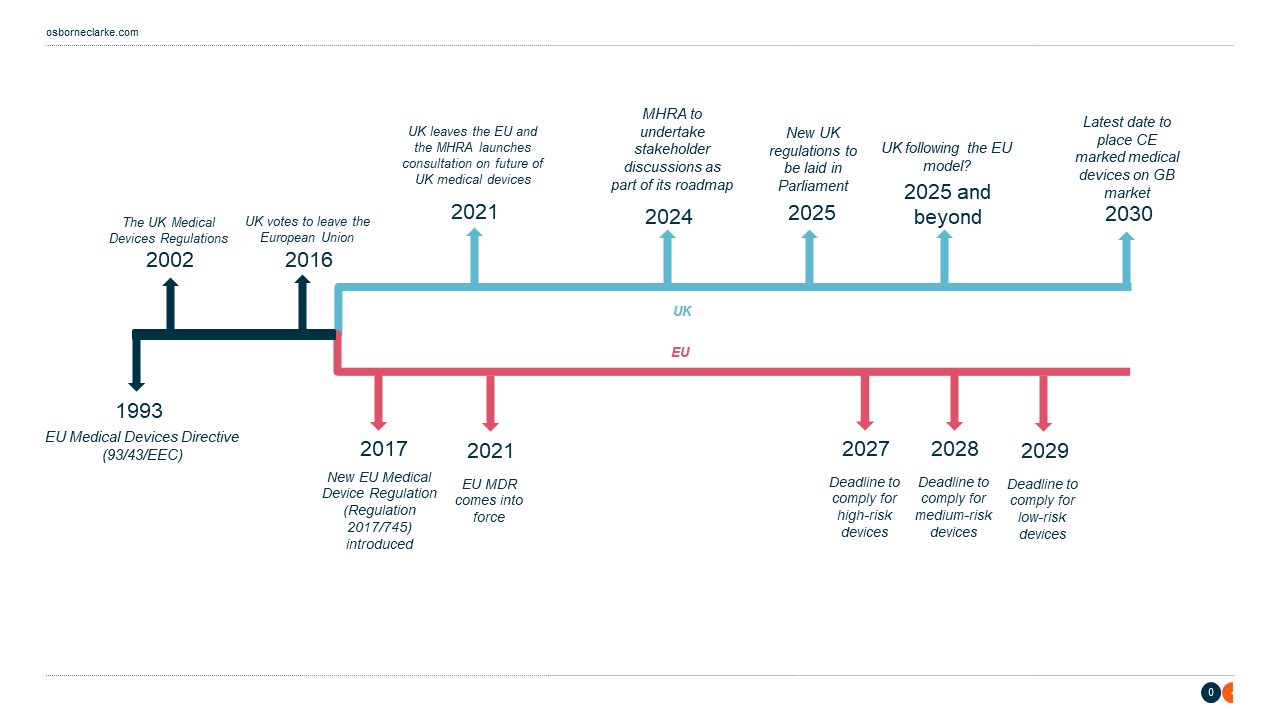
The roadmap outlines the key action points taken between 2021 and 2023, as well as numerous stakeholder discussions that will take place throughout 2024, with plans for priority measures to be in place this year and future core regulations to be introduced during 2025.
The MHRA intends to establish UK regulations that promote international harmonisation and prioritise patient-centred and proportionate rules, including processes to recognise devices approved by overseas regulators and measures to bring requirements in line with EU rules.
In the meantime, the latest government extension allowing manufacturers to place CE- marked medical devices on the market in Great Britain means that a product compliant with the EU MDR can be placed on the Great Britain market up until June 2030.
Osborne Clarke comment
Industry will be looking forward to receiving more information on proposed new UK medical devices regulations as the MHRA begins to progress with its roadmap. Draft regulations have not yet been published; however, it appears that the MHRA is considering an approach for UK regulations that would broadly mirror the EU model.
Taking into account the MHRA's statements to date, as well as the latest extensions allowing CE-marked products onto the market in Great Britain, medical device manufacturers may find that compliance with the EU model is an approach that will facilitate compliance with future medical devices regulations in the UK.
Manufacturers should monitor both available extensions for compliance with the MDR in the EU, as well as developments in the UK, as the MHRA adds detail to its proposals.
Jamie Roberts, a Trainee Solicitor with Osborne Clarke, contributed to this article.